




Czynnik genetyczny występuje u ok. 30% niepłodnych par (w tym większość stanowią przypadki tzw. niepłodności idiopatycznej).
Kariotyp
Kiedy myślimy o genetyce, przypomina się nam obraz chromosomów, o których uczyliśmy się w szkole. Każdy człowiek posiada 23 pary chromosomów, w tym jedną parę chromosomów płci: XX lub XY. Jeśli chcemy sprawdzić, czy wszystkie chromosomy są „na swoim miejscu”, wykonujemy badanie kariotypu. Należy podkreślić, że tylko 0,5% populacji ma nieprawidłowy kariotyp. Zapytacie więc, po co wykonywać badanie kariotypu, które jest jednym z droższych badań, skoro ryzyko jakiejkolwiek wady wynosi 0,5%?
Po pierwsze, w populacji osób niepłodnych, wady genetyczne zdarzają się nieco częściej: u niepłodnych mężczyzn w 1,1-7,2%, a u niepłodnych kobiet w nawet 10% przypadków (doi: 10.1515/bjmg-2015-0002).
Według rekomendacji „Diagnostyka i leczenie niepłodności – rekomendacje Polskiego Towarzystwa Medycyny Rozrodu i Embriologii oraz Polskiego Towarzystwa Ginekologów i Położników” z 2018 roku badanie kariotypu jest zalecane:
– u pacjentów z azoospermią lub u których koncentracja plemników w nasieniu nie przekracza 5mln/ml
– jeżeli w wywiadzie uzyskano informację o poronieniach nawracających lub jeżeli materiał genetyczny z poronienia wskazuje na możliwość nosicielstwa zmiany genetycznej u rodziców.
I to właśnie nieprawidłowy wynik badania nasienia oraz nawracające poronienia są najczęstszymi przypadkami, gdy w kariotypie mogą wystąpić jakieś wady.
Istnieją dwa podejścia: w pierwszym wykluczamy tę przyczynę na początku leczenia wykonując badanie kariotypu u obojga partnerów (a gdy wszystko w porządku, szukamy innych przyczyn), a w drugim wykonujemy badanie kariotypu zgodnie ze wspomnianymi rekomendacjami PTMRiE i PTGiP. Ja zdecydowanie jestem „za” pierwszym podejściem, jeśli tylko pacjenci mają możliwości finansowe do wnikliwej diagnostyki. Dzięki temu możemy uniknąć ryzyka urodzenia chorego dziecka poprzez zastosowanie diagnostyki genetycznej zarodków w przypadku wykrycia nieprawidłowości, a jeśli wszystko będzie w porządku… No właśnie – często wśród pacjentów panuje przekonanie, że jeśli kariotypy są w porządku, to nie ma ryzyka urodzenia chorego dziecka. Niestety nie jest to prawdą. Proces gametogenezy, czyli powstawania komórek jajowych i plemników, jest „narażony” na błędy. Produkcja oocytów i plemników to nic innego, jak podziały komórek (ze szkoły pamiętamy mejozę i mitozę). W czasie podziału komórki, materiał genetyczny musi zostać rozdzielony idealnie po równo do komórek potomnych. Niestety, rozdział ten często zachodzi nieprawidłowo (częściej u kobiet starszych, u których mechanizmy rozdzielania chromosomów mogą być już „zepsute”). Dlatego mimo, że kobieta posiada 23 pary chromosomów, w jej komórce jajowej może funkcjonować zmieniona liczba chromosomów. Stąd tak ważne jest, aby nie sugerować się kariotypem, tylko wykonać badanie genetyczne zarodków, jeśli pacjentka jest po 35. roku życia lub występują inne wskazania.
Kiedy można podejrzewać, że przyczyną niepłodności jest czynnik genetyczny (u kobiet i mężczyzn)?
– przedwcześnie wygasająca rezerwa jajnikowa (POI/POF)
– wrodzony hipogonadyzm hipogonadotropowy
– wady rozwojowe narządów płciowych
– pierwotny brak miesiączki
– nieprawidłowy rozwój III-rzędowych cech płciowych (specyficzne owłosienie ciała, specyficzne proporcje budowy ciała, barwa głosu, biust u kobiet, jabłko Adama u mężczyzn)
– nawracające poronienia
– azoospermia (brak plemników), oligozoospermia (gdy koncentracja plemników jest niższa niż 5 mln/ml)
Jakie wady genetyczne występują u niepłodnych mężczyzn?
Ponad 15% przypadków męskiej niepłodności może być spowodowanych czynnikiem genetycznym.
W przypadku nieprawidłowych parametrów nasienia, lekarz zleca najczęściej wykonanie badań kariotypu, AZF i CFTR:
- Najczęściej występujące wady kariotypu to zespół Klinefeltera (47, XXY lub 46, XY/47, XXY) oraz translokacje robertsonowskie. O ile zespół Klinefeltera charakteryzuje zestaw specyficznych cech (wysoki wzrost, przerost sutków, brak owłosienia na twarzy i ciele, małe jądra, azoospermia, wzrost FSH i LH oraz spadek testosteronu w surowicy krwi), to w przypadku zrównoważonej translokacji, pacjent będzie zupełnie zdrowy. Translokacja zrównoważona to przeniesienie fragmentu jednego chromosomu na inny. Liczba chromosomów się zgadza, pacjent nie ma żadnych objawów, a jedynie nieprawidłowo zachodzi produkcja plemników, co może prowadzić do obecności translokacji niezrównoważonej u potomstwa.

- AZF to region na chromosomie Y, którego delecje mogą powodować:
– całkowity brak plemników (jeśli delecja obejmuje region AZFa)
– zatrzymanie dojrzewania plemników (jeśli delecja obejmuje region AZFb)
– ciężką oligozoospermię lub azoospermię (jeśli delecja obejmuje region AZFc)
- CFTR to mutacja odpowiedzialna za wrodzony obustronny brak nasieniowodów, co powoduje blokadę transportu plemników i w rezultacie azoospermię. W przypadku obecności nieprawidłowego wariantu należy ZAWSZE wykonać badanie CFTR u partnerki.
Dodatkowo, istnieje ryzyko mutacji w wielu genach związanych z męską płodnością. W 2019 roku ukazała się publikacja (doi:10.1093/humrep/dez022) która porusza temat jednogenowych mutacji wpływających na produkcję hormonów, nieprawidłowe parametry nasienia (teratozoospermię, oligozoospermię, asthenozoospermię), proces spermatogenezy i wiele innych. Znanych jest ponad 600 takich genów. Przykładowo:
- Geny PRM1 i PRM2 kodujące protaminy, czyli białka jądrowe plemników – mutacje w tych genach mogą prowadzić do nieprawidłowej kondensacji chromatyny (nieprawidłowe dojrzewanie plemników), co przyczynia się np. do niewyjaśnionych poronień u partnerki
- Gen AURKC odpowiada za prawidłowe funkcjonowanie białka uczestniczącego w formowaniu wrzeciona podziałowego i segregacji chromosomów – mutacje w tym genie mogą prowadzić do błędnego rozdziału materiału genetycznego w czasie spermatogenezy, powstawania ogromnych główek plemników (makrozoospermia) i aneuploidii w plemnikach. DOI:10.1016/j.androl.2019.04.004
- Geny DNAI1 i DNAH5 odpowiadają za prawidłową budowę dyneiny (białko znajdujące się w witce plemnika i odpowiadające za ruch witki) – ich mutacje mogą prowadzić do zaburzenia budowy wewnętrznej witki plemnika, co powoduje asthenozoospermię lub nawet całkowity brak ruchomych plemników (zespół Kartagenera, pierwotna dyskineza rzęsek). doi: 10.4103/1008-682X.122192
- MMAF – multiple morphological anomalies of the flagella – czyli wielokrotne anomalie morfologiczne witek plemników – jest to rodzaj teratozoospermii polegający na obecności wad witek w ogromnym odsetku plemników (ponad 80% witek jest krótkich, nieregularnych, zrolowanych, połamanych lub są same główki bez witek), jednocześnie plemniki te wykazują brak ruchu, zatem nieruchoma jest większość lub nawet 100% plemników. Według naukowców przyczyna występowania MMAF może być heterogeniczna: u 30% badanych pacjentów wystąpiła mutacja genu DNAH1, ale u podłoża mogą leżeć mutacje innych genów: CFAP44, CFAP43, AK7, CFAP69, CEP135, AKAP3, AKAP4 i zapewne także jeszcze kilku jak dotąd niezweryfikowanych. DOI:https://doi.org/10.1016/j.rbmo.2017.12.005 doi: 10.4103/aja.aja_53_19
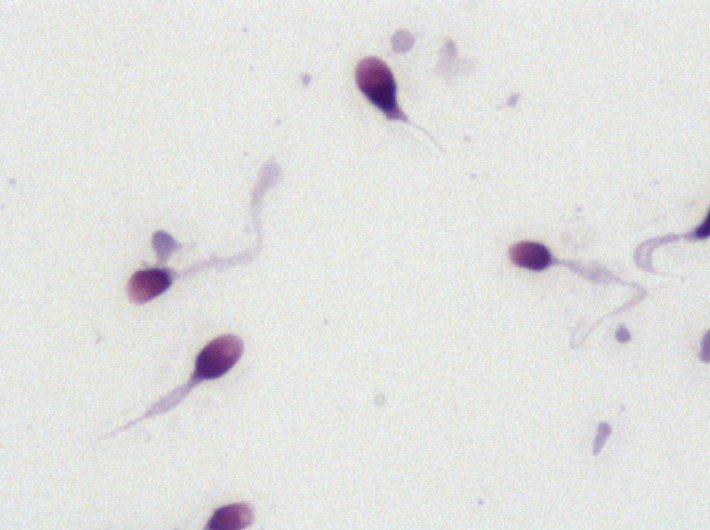
Podsumowując, standardowe badania genetyczne dla mężczyzn to badanie AZF, CFTR oraz kariotyp. Można także zweryfikować gen ANXA5, który może odgrywać rolę w nawracających poronieniach. Co do innych genów, to nie wszystkie z nich zostały bezpośrednio powiązane z wpływem na męską płodność. Przykładowo, istnieje kilka potencjalnych genów, które mogą powodować globozoospermię (rodzaj teratozoospermii), jednak np. mutację PICK1 posiadała tylko jedna z badanych osób i naukowcy nie są w stanie wskazać jednoznacznie, że mutacja X na sto procent wywołuje objaw Y. Nie ma zatem sensu jej badać, skoro nie znamy ostatecznego rezultatu. Warto też pamiętać, że nie istnieje leczenie przyczynowe – nie ma możliwości naprawy mutacji genetycznej (przykładowo: jeśli pacjent ma globozoospermię, nie ma możliwości naprawy genu, który ją wywołuje. Leczenie polega na zapłodnieniu pozaustrojowym ICSI lub skorzystaniu z banku nasienia, co i tak wykonuje się standardowo u tych pacjentów).
Jakie wady genetyczne występują u niepłodnych kobiet?
Wady w kariotypie u kobiet mogą prowadzić nie tylko do zaburzeń płodności takich jak hipogonadyzm hipogonadotropowy czy brak miesiączek (delecje na chromosomie X), ale także do poronień.
Zaburzenia genetyczne i ich wpływ na płodność u kobiet (doi: 10.1093/biolre/ioz084):
- Ok. 1-2% kobiet doświadcza POI (przedwczesnego wygasania czynności jajników), który polega na braku miesiączki, niepłodności, obniżonym poziomie estrogenów, podwyższonym poziomie FSH i LH, zwiększonym ryzyku osteoporozy i chorób naczyniowych. W 10% u podłoża POI może leżeć przyczyna genetyczna: zespół Turnera, delecje, duplikacje oraz zrównoważone i niezrównoważone rearanżacje chromosomu X i autosomów.
- Zespół łamliwego chromosomu X – mutacja w genie FMR1 może także prowadzić do przedwczesnej niewydolności jajników POI.
- Delecje, duplikacje, inwersje, translokacje między chromosomem X i autosomami oraz mutacje pojedynczych genów u 5-6% kobiet mogą powodować nawracające poronienia. Co ciekawe, mutacje genów na chromosomie X: BCOR, EBP, FLNA, HCCS, IKBKG, MECP2, OFD1, OTC i REP1 mogą prowadzić do poronień męskich płodów.
- U podstaw przedwczesnej niewydolności jajników, pierwotnego braku miesiączki, dysgenezji gonad (brak prawidłowego rozwoju jajników), hipogonadyzmu hipogonadotropowego i niepłodności kobiet (zaburzenia oogenezy, nieprawidłowe podziały komórkowe, funkcjonowanie mitochondriów lub gospodarka hormonalna) mogą leżeć mutacje wielu genów (znanych jest ponad 350). Podobnie zatrzymanie rozwoju zarodków może mieć także przyczynę genetyczną.
- Mutacje genetyczne w genach FSHR, BMP15 i LHCGR są związane z zaburzeniami hormonalnymi w zespole policystycznych jajników (PCOS). Także mutacja genu dla receptora LH może występować u kobiet z zespołem pustego pęcherzyka.
- Mutacje glikoprotein ZP1, ZP2, ZP3 i ZP4, które znajdują się na powierzchni oocytu i odpowiadają za wiązanie plemnika do komórki jajowej, mogą prowadzić do: braku osłony przejrzystej na oocytach, nieprawidłowej osłony przejrzystej, zespołu pustego pęcherzyka, degeneracji oocytów, zaburzeń w dojrzewaniu oocytu i wreszcie, braku zapłodnienia.
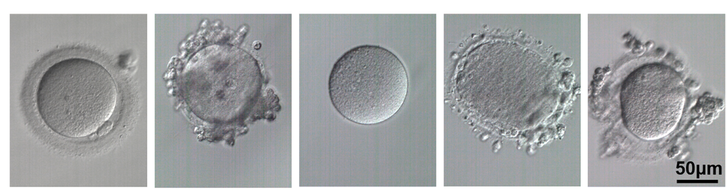
- Mutacje wielu genów powodują zatrzymanie rozwoju zarodka. Zwykle do 70% zarodków rozwija się do etapu blastocysty. Pozostałe zarodki zatrzymują się w rozwoju, głównie z powodu aneuploidii, ale także, jak wykazali naukowcy, z powodu nieprawidłowych wariantów genów u matki. Te najważniejsze geny to: PADI6, PATL2, TLE6, NLRP2, NLRP5, WEE2, TUBB8, OOEP, NLRP5, TLE6, KHDC3L, TLE6, NLRP5. Co ciekawe, nieprawidłowy wariant genu KHDC3L jest związany z nawracającymi przypadkami zaśniadu groniastego oraz zatrzymaniem rozwoju zarodków na etapie moruli. Mutacja genu WEE2 powoduje z kolei brak zapłodnień.
- U ok. 3% kobiet występują zaburzenia w budowie i funkcjonowaniu gonad: agenezja, atrezja, nieprawidłowy rozwój jajników, macicy, szyjki macicy lub pochwy. Wady budowy macicy mogą powstać w wyniku mutacji genu HOXA13 (hand-foot-genital syndrome) oraz genów HNF1B, LHX1, WNT4, WNT7A i WNT9B (np. Zespół Mayer-Rokitansky-Küster-Hauser, który charakteryzuje się agenezją pochwy i macicy oraz pierwotnym brakiem miesiączki – u tych kobiet jajniki funkcjonują prawidłowo, stąd jedyną możliwością na posiadanie potomstwa jest skorzystanie z surogatki)
Najczęstszymi genetycznymi przyczynami poronień nawracających są obecne u kobiety:
– translokacje zrównoważone (59%)
– translokacje Robertsonowskie (27%)
– inwersje (9%)
– aneuploidie chromosomów płci (4%)
– nadliczbowe chromosomy (1%)
Poronienie to utrata ciąży przed 22. tygodniem. O poronieniach nawracających mówimy, gdy wystąpią 2 lub więcej utraty ciąży. Poronienie należy też odróżnić od ciąży biochemicznej – ciąża biochemiczna to dodatni wynik testu beta HCG, który nie utrzymał się i poziom hormonu HCG spadł zanim stwierdzona została akcja serca płodu.
Warto pamiętać, że główną przyczyną wszystkich poronień są wady genetyczne płodu (50-60%).
W poronieniach nawracających rolę mogą odgrywać także mutacje w genach: aneksyny V (ANXA5), PAI-I, (SERPINE1), czynnika V Leiden (F5) oraz genu protrombiny (F2), które prowadzą do zwiększonego poziomu krzepliwości krwi, co wiąże się z podwyższonym ryzykiem chorób zakrzepowo-zatorowych. Standardowo wykonuje się badania genetyczne dla powyższych genów w przypadku nawracających powikłań położniczych.
Problem genetycznej przyczyny niepłodności jest złożony, a przede wszystkim, nie do końca poznany. Ze względu na brak możliwości naprawy wadliwych genów, rozpoznanie mutacji genetycznej u pacjenta stawia przed lekarzem wyzwanie, aby szukać innych sposobów terapii.